1. Arzneimittel-Zulassung und frühe Nutzenbewertung in Deutschland
Möchte ein Pharmahersteller in Deutschland ein neues verschreibungspflichtiges Arzneimittel auf den Markt bringen, muss er zwei Verfahren durchlaufen:
- die Zulassung, um sein Mittel in Deutschland verkaufen zu dürfen und erstattet zu bekommen, und
- die frühe Nutzenbewertung, anhand derer ermittelt wird, wie hoch die Kostenübernahme durch die Krankenkassen nach den ersten sechs Monaten ausfällt.
Die Zulassung
Möchte ein pharmazeutisches Unternehmen oder eine Forschergruppe ein Arzneimittel auf den Markt bringen oder der Öffentlichkeit zugänglich machen, muss zunächst eine Zulassung beantragt werden, also eine staatliche Erlaubnis, dieses Mittel in Verkehr zu bringen.
Eine Zulassung ist nicht erforderlich, wenn das Mittel zu einer der folgenden Gruppen gehört (siehe 4. Besondere Therapierichtungen und traditionelle Arzneimittel):
- Lebensmittel und Nahrungsergänzungsmittel,
- kosmetische Mittel oder
- homöopathische und andere Mittel der besonderen Therapierichtungen.
Je nachdem, wo das Arzneimittel vertrieben werden soll, gibt es innerhalb der EU vier Verfahrenswege, eine Zulassung zu beantragen:
- das nationale Verfahren, um das Mittel nur in Deutschland zu vertreiben,
- zwei dezentrale Verfahren, um ein Mittel in mehreren Ländern der EU zu vertreiben und
- das zentrale Verfahren, um ein Mittel in allen Ländern des Europäischen Wirtschaftsraums (EWR) zu vertreiben.
Bis 1995 waren die nationalen Verfahren die gängigen Wege, um ein Arzneimittel in einem Land zu vertreiben. Sie spielen indes seit der Einführung des zentralen Verfahrens der EU eine weniger bedeutende Rolle. Hinzu kommt: Gehört ein neues Arzneimittel zu einer bestimmten Gruppe von Medikamenten, ist der zentrale Weg sogar zwingend vorgeschrieben. Dazu zählen neue Arzneimittel gegen Krebs, neurodegenerative Erkrankungen, Diabetes, HIV, Viruserkrankungen generell, Autoimmunerkrankungen und andere Immunschwächen sowie seltene Erkrankungen.
Möchte ein Hersteller ein Arzneimittel, das nicht zu dieser Gruppe zählt, nur in Deutschland vertreiben, wendet er sich mit den Zulassungsunterlagen an die entsprechende nationale Behörde, das Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) oder das Paul-Ehrlich-Institut (siehe 5. Die Institutionen).
Möchte ein Hersteller sein Arzneimittel außer in Deutschland auch in einigen anderen Ländern innerhalb der EU vertreiben, erreicht er dies über eines von zwei dezentralen Verfahren: das eigentliche dezentralisierte Verfahren (Decentralised Procedure = DCP), wenn das Arzneimittel noch in keinem Land zugelassen ist, oder das Verfahren der gegenseitigen Anerkennung (Mutual Recognition Procedure = MRP), wenn das Mittel bereits in einem EU-Land zugelassen ist.
Im DCP wird ein Antrag auf Zulassung in den gewünschten Ländern mit einem identischen Dossier gestellt. Im MRP prüfen die Zulassungsstellen der anderen Länder die Zulassungsunterlagen des Landes, das die Zulassung bereits erteilt hat, und stimmen der Zulassung auf dieser Grundlage zu.
Möchte ein Hersteller sein Arzneimittel in der gesamten EU und den Ländern des EWR (Liechtenstein, Island, Norwegen) vertreiben, wendet er sich an die Europäische Arzneimittelagentur (EMA) für das zentrale Zulassungsverfahren.
Abseits dieser regulären Zulassungsverfahren gibt es alternative Wege für schnellere Zulassungen (etwa in Notzeiten) oder Zulassungen für besondere Umstände (etwa für seltene Erkrankungen).
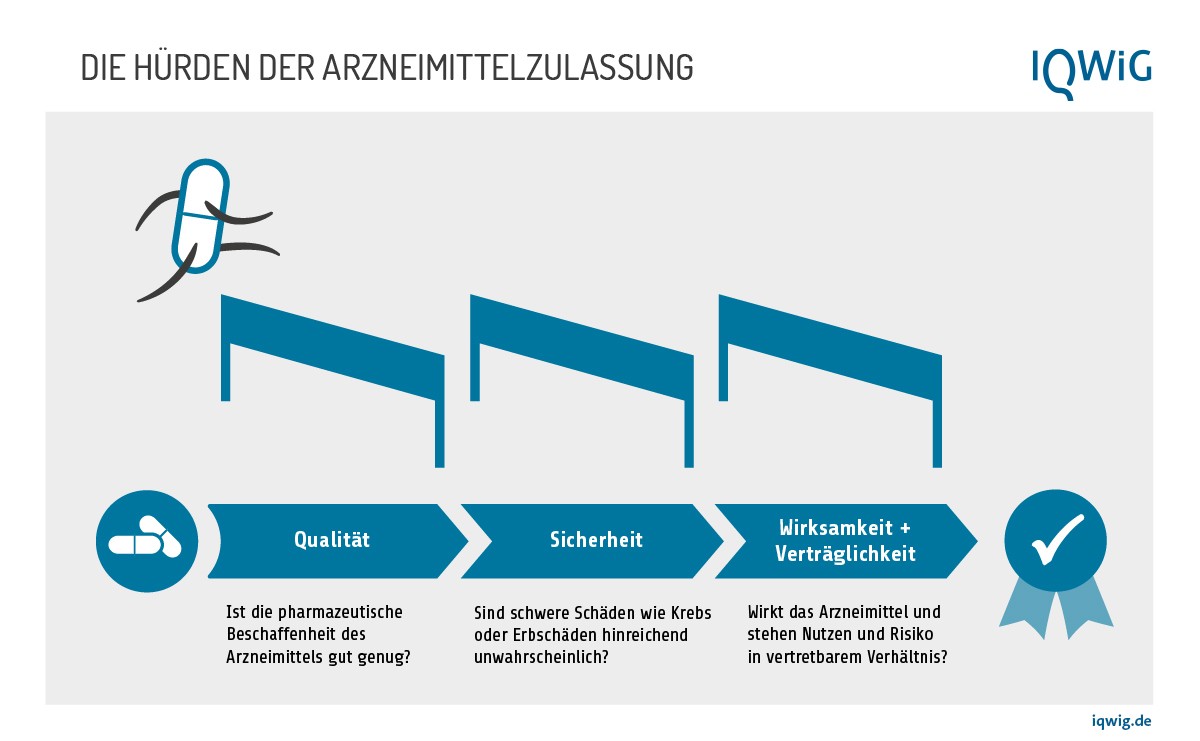
Was wird geprüft? In der Zulassung muss das Präparat drei Hürden nehmen, weil folgende Parameter eines Wirkstoffs geprüft werden:
- die pharmazeutische Qualität,
- die Sicherheit und
- die Wirksamkeit und Verträglichkeit.
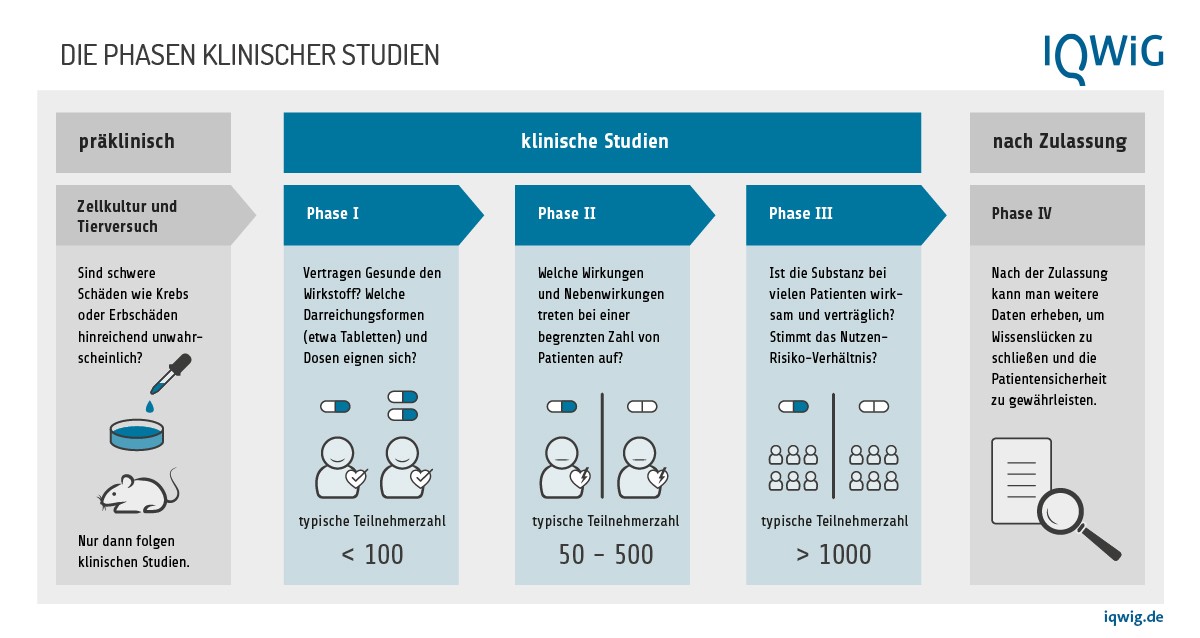
Für die Überprüfung der pharmazeutischen Qualität gibt es vorgeschriebene Tests, die durchgeführt werden müssen. Sicherheit, Wirksamkeit und Verträglichkeit werden in Studien ermittelt, angefangen von einfachen Laboruntersuchungen im Reagenzglas über Tierversuche und klinische Studien mit zunächst nur wenigen Teilnehmerinnen und Teilnehmern bis hin zu großen Untersuchungen mit vielen hundert oder tausend Teilnehmern und Kontrollgruppen (Phase 1 bis 3).
Leider ist der Ausdruck Sicherheit oder Unbedenklichkeit, englisch safety, in den Gesetzen, Verordnungen und Richtlinien zum Thema doppelt belegt. Wir verstehen hier unter "Sicherheit" den Ausschluss schädlicher Substanzen in der vorklinischen Überprüfung und nennen die Vertretbarkeit von Nebenwirkungen, die in klinischen Studien ermittelt werden, "Verträglichkeit".
Wenn die Studien zeigen, dass der Nutzen die Risiken überwiegt, erfolgt die Zulassung. Ob ein Medikament besser wirkt oder zum Beispiel weniger Nebenwirkungen hat als andere, die bereits auf dem Markt sind, spielt dagegen bei der Zulassung keine Rolle. Es genügt, wenn das Mittel in kontrollierten Studien besser ist als ein Placebo (ein Scheinmedikament) oder zumindest nicht schlechter ist als ein Standardpräparat, das nachweislich besser als Placebo ist. Das Ausmaß des Nutzens wird erst im zweiten Schritt, der frühen Nutzenbewertung, wichtig. Dann wird in Deutschland überprüft, ob das neue Mittel den Patientinnen und Patienten besser hilft als eine Standardtherapie, also einen Zusatznutzen hat.
Ein geprüftes Arzneimittel wird für fünf Jahre zugelassen. Danach kann ein Hersteller eine Verlängerung beantragen, die unbegrenzt gilt, sofern es keine Sicherheitsbedenken gibt. Die Kosten für das Arzneimittel werden mit dem Beginn der Zulassung von den gesetzlichen Krankenkassen übernommen. Der Preis für das Mittel wird für die ersten sechs Monate alleine vom Hersteller festgesetzt.
Die frühe Nutzenbewertung

Neue Arzneimittel sind in der Regel teurer als die auf dem Markt etablierten Präparate; Hersteller begründen dies mit den hohen Entwicklungskosten und einer besseren Leistung dieser neuen Mittel im Vergleich zu bestehenden Präparaten. Doch die Budgets der Gesundheitssysteme sind begrenzt und es gibt seit jeher begründete Zweifel, ob neue Arzneimittel per se besser sind als die auf dem Markt befindlichen. Daher wurde in Deutschland auf Grundlage des Arzneimittelmarktneuordnungsgesetzes (AMNOG) ab dem 1. Januar 2011 ein Verfahren eingeführt, in dem der tatsächlich für die Patienten relevante Nutzen des neuen Mittels mit dem eines etablierten Präparats oder einer etablierten Behandlungsstrategie verglichen wird.
Auf Grundlage dieses Vergleichs wird dann entschieden, welchen Preis bzw. Betrag die Krankenkassen übernehmen. Diese Nutzenbewertung wird als "frühe" Nutzenbewertung bezeichnet, weil sie unmittelbar nach der Markteinführung in Deutschland durchgeführt wird.
Spätestens zum Zeitpunkt des erstmaligen Inverkehrbringens in Deutschland muss der Hersteller für die frühe Nutzenbewertung ein umfassendes Dossier an den Gemeinsamen Bundesausschuss (G-BA) übergeben. In diesem Dossier soll der Hersteller den patientenrelevanten Zusatznutzen des neuen Arzneimittels im Vergleich mit einem etablierten Mittel belegen. Dies geschieht auf Basis von Daten aus veröffentlichten und unveröffentlichten klinischen Studien mit dem neuen Arzneimittel. Darüber hinaus gibt es im Dossier Schätzungen für die jährliche Zahl der Patienten, die potenziell von dem neuen Medikament profitieren, und die prognostizierten jährlichen Arzneimittelkosten.

Die Unterlagen zum patientenrelevanten Zusatznutzen übergibt der G-BA an das IQWiG, das anhand dieser Unterlagen einen Vergleich zwischen neuem und etabliertem Mittel durchführt. Das gesamte Verfahren folgt einem strengen Zeitplan, sodass nach einem Jahr, spätestens aber nach 15 Monaten der Prozess abgeschlossen sein muss.
Abhängig davon, welchen Zusatznutzen der G-BA dem neuen Wirkstoff attestiert (wie zum Beispiel eine verringerte Sterblichkeit oder eine verbesserte Lebensqualität), ergibt sich dann in den Preisverhandlungen zwischen Krankenkassen und Herstellern, zu welchem Preis der Hersteller sein neues Arzneimittel in Deutschland erstattet bekommt. Das Ausmaß des Zusatznutzens ist aufsteigend gestaffelt nach
- gering (niedrigstes Ausmaß),
- beträchtlich (mittleres Ausmaß) und
- erheblich (höchstmögliches Ausmaß).
Ergibt die Bewertung keinen Zusatznutzen (entweder, weil er laut Studien nicht belegt ist, oder weil die Daten gar nicht ausreichen für einen aussagekräftigen Vergleich), kann das Mittel zwar auf dem Markt bleiben. Es wird aber nur noch mit dem für diese Arzneimittelgruppe bestimmten Festbetrag erstattet bzw. einem Betrag für ein Jahr Therapie, der nicht höher sein darf als für eine etablierte Vergleichstherapie. Generell gilt also: Ein neues Arzneimittel ohne zusätzlichen Nutzen darf nicht mehr kosten als die Standardtherapie.
Dass für ein Produkt kein Zusatznutzen belegt ist, muss nicht bedeuten, dass der neue Preis zwangsläufig unter dem vom Hersteller zuvor festgesetzten Preis liegt, wenn das Vergleichsprodukt aufgrund eines Patentschutzes bereits einen hohen Preis erzielen konnte.
Laufende und in der Vergangenheit durchgeführte Nutzenbewertungen sind auf der Website des G-BA dokumentiert.
Neue Arzneimittel: Zulassung, Nutzenbewertung, Erstattung
1. Arzneimittel-Zulassung und frühe Nutzenbewertung in Deutschland
2. Arzneimittel-Zulassung und -Nutzenbewertung in anderen Ländern
3. Das AMNOG-Verfahren: mehr als nur Kostenkontrolle
4. Besondere Therapierichtungen und traditionelle Arzneimittel

