1. Drug approval and early benefit assessment in Germany
If a pharmaceutical company wants to market a new prescription drug in Germany, the drug is subject to two procedures:
- drug approval (also called marketing authorization) in order to be allowed to sell the drug in Germany, with coverage of costs by the statutory health insurance (SHI) funds, and
- early benefit assessment, which is used to determine the extent of coverage by the SHI funds after the first six months.
Drug approval
If a pharmaceutical company or a group of researchers wishes to market a drug or make it available to the public, it must first apply for approval, i.e. a regulatory permit to place this drug on the market. Approval is not required if the product belongs to one of the following groups (see 4. Particular therapeutic systems and traditional drugs):
- food and dietary supplements,
- cosmetic products or
- homeopathic and other remedies of so-called "particular therapeutic systems"
Depending on where the drug is to be marketed, there are four procedural routes within the EU to apply for approval:
- the national procedure - to market a drug only in Germany,
- two decentralized procedures - to market a drug in some EU countries, and
- the centralized procedure - to market a drug in all countries of the European Economic Area (EEA).
Until 1995, national procedures were the most common ways to market a drug in a country. Since the introduction of the centralized EU procedure, however, they have played a less important role. In addition, if a new drug belongs to a certain group of drugs, the centralized route is mandatory. This applies to new drugs for cancer, neurodegenerative diseases, diabetes, HIV, viral diseases in general, autoimmune diseases, other immune deficiencies, and rare diseases.
If a company wishes to market a drug that does not belong to the above group only in Germany, it submits the documents supporting approval to the corresponding national authority, the Federal Institute for Drugs and Medical Devices (BfArM) or the Paul Ehrlich Institute (see "5. The institutions").
If, besides in Germany, a pharmaceutical company wants to market its drug in some other countries within the EU, one of two decentralized procedures apply: the Decentralized Procedure (DCP) if the drug has not yet been approved in any EU country or the Mutual Recognition Procedure (MRP) if the drug has already been approved in an EU country.
In the DCP, an application for approval is submitted in the target countries with an identical dossier. In the MRP, the regulatory authorities of the other countries review the regulatory dossier of the country that has already granted approval and grant approval on this basis.
If a pharmaceutical company wants to market its drug in the whole EU and the other countries of the EEA (Liechtenstein, Iceland, Norway), it contacts the European Medicines Agency (EMA) with regard to the central authorization procedure.
Beyond these regular approval procedures, there are alternative pathways for faster approvals (e.g. emergency approvals) or approvals for special circumstances (e.g. for rare diseases).
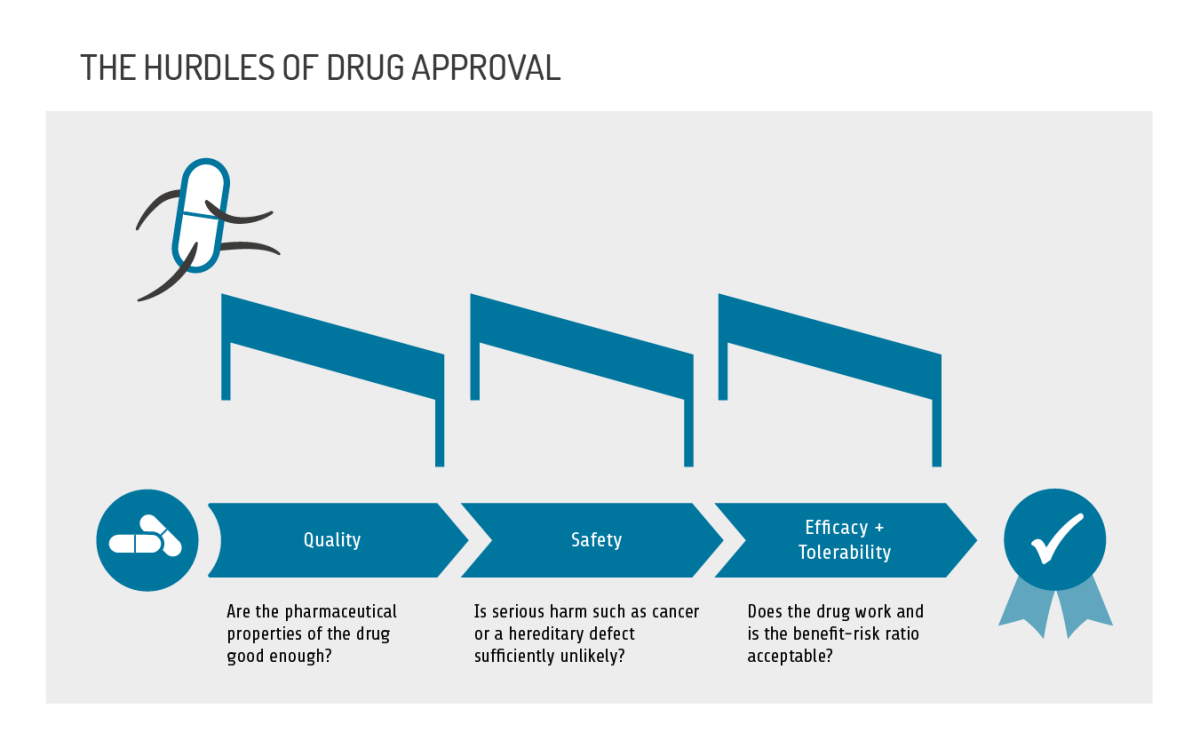
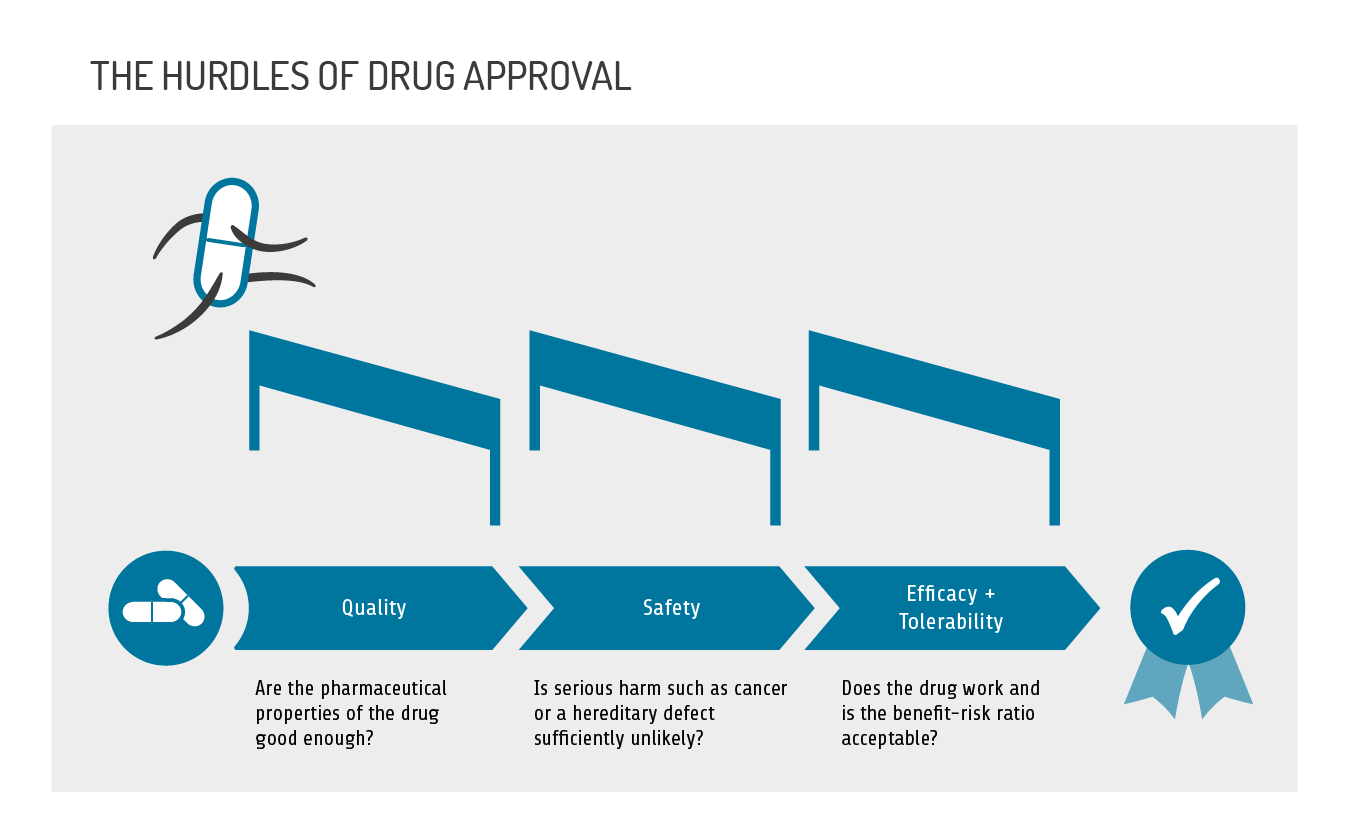
What is examined? In the approval process, the drug has to pass three hurdles, as the following features of a drug are examined:
- pharmaceutical quality
- safety
- efficacy and tolerability.
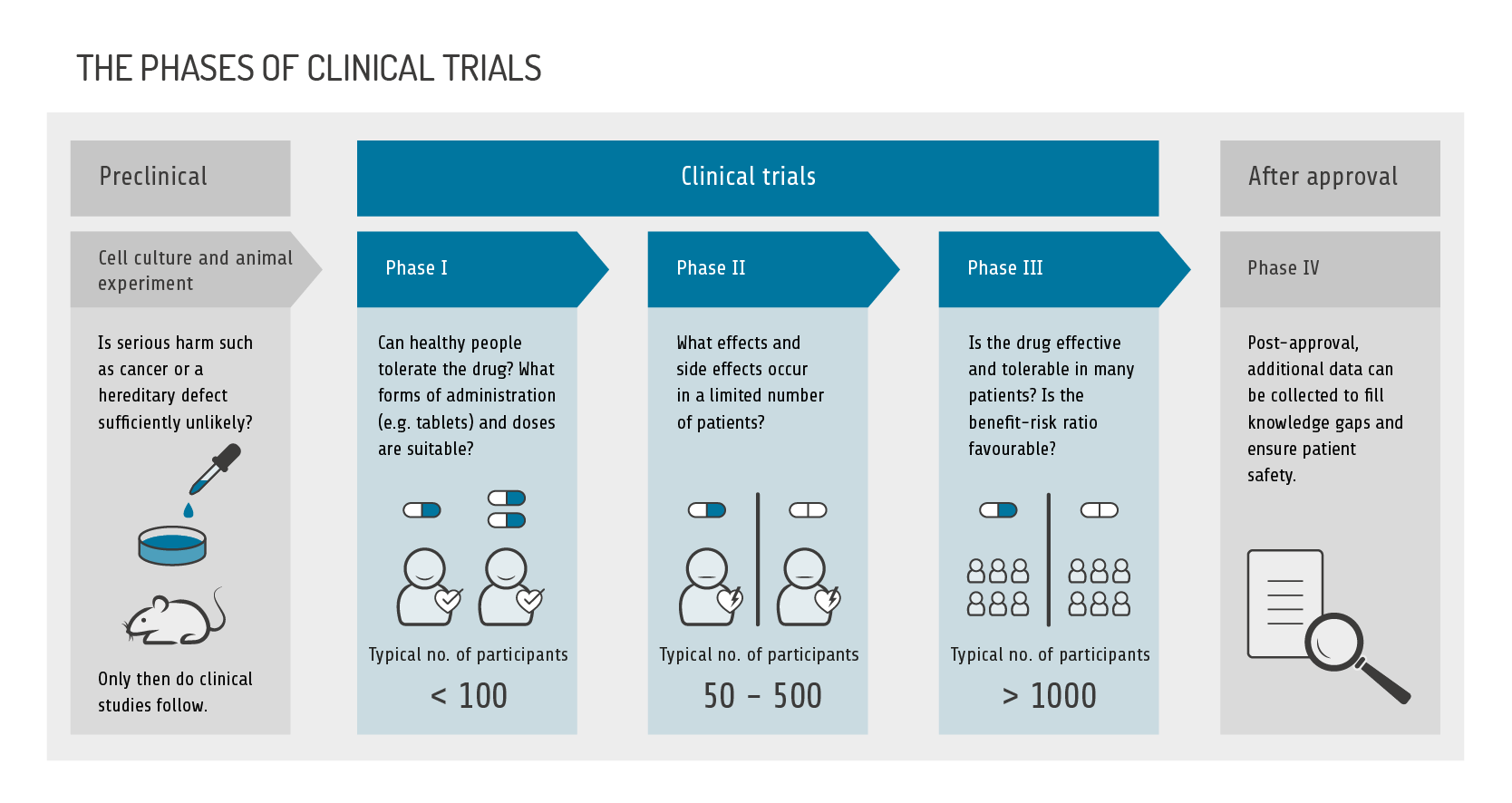
Mandatory tests must be performed to verify pharmaceutical quality. Safety, efficacy and tolerability are determined in studies, ranging from simple laboratory tests in a test tube, to animal experiments, to clinical trials with initially only a few participants, and finally to large clinical trials with many hundreds or thousands of participants in test and control groups (phases 1 to 3).
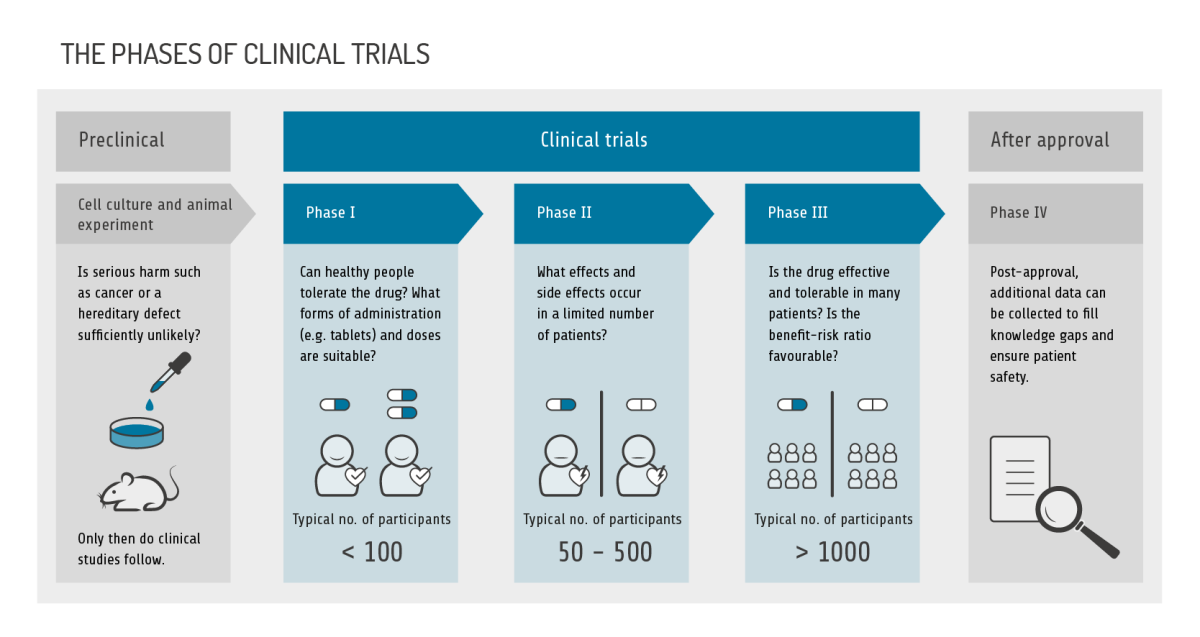
The definition of safety is inconsistent in the various national and international laws and regulations. In the context described here, we understand "safety" to mean the elimination of harmful drugs in preclinical testing and call the acceptability of side effects shown in clinical trials "tolerability".
If the studies show that the benefits outweigh the risks, approval is granted. Whether a drug is more effective or, for example, has fewer side effects than other drugs already on the market plays no role in the approval process. It is sufficient if the drug is better than a placebo (a dummy drug) in controlled trials, or at least is no worse than a standard drug that has been shown to be better than placebo. The extent of benefit only becomes important in the second step, the early benefit assessment. Then, in Germany, it is examined whether the new drug helps patients better than the standard treatment, i.e. whether it has an added benefit.
A drug that passes the approval procedure is approved for five years. After that, the pharmaceutical company can apply for an extension, which is unlimited, provided there are no safety concerns. The cost of the drug is covered by the SHI funds from the time of approval. For the first six months, the price of the drug is set by the pharmaceutical company alone.
Early benefit assessment

New drugs are usually more expensive than drugs already established on the market; pharmaceutical companies usually justify this with the high development costs and better performance of these new drugs compared to the existing ones. However, the budgets of health care systems are limited and there have always been reasonable doubts as to whether new drugs are per se better than those on the market. Therefore, on 1 January 2011, a procedure was introduced in Germany based on the Act on the Reform of the Market for Medicinal Products (AMNOG), in which the actual patient-relevant benefit of a new drug is compared with that of an established drug or treatment strategy.
On the basis of this comparison, a decision is then made as to which price will be covered by the SHI funds. This benefit assessment is referred to as an "early" benefit assessment because it is conducted immediately after market launch in Germany.
For the early benefit assessment, the pharmaceutical company must submit a comprehensive dossier to the Federal Joint Committee (G-BA) no later than the time of first marketing in Germany. In this dossier, the pharmaceutical company must demonstrate the patient-relevant added benefit of the new drug in comparison with an established drug. This is done based on data from published and unpublished clinical trials on the new drug. In addition, the dossier contains estimates on the annual number of patients potentially benefiting from the new drug as well as the projected annual drug costs.

The G-BA transfers the dossier to IQWiG, which uses these documents to conduct a comparison between the new and established drug. The entire process follows a strict timetable; the process must be completed after one year, or after 15 months at the latest.
Depending on which added benefit the G-BA assigns to the new drug (e.g. reduced mortality or improved quality of life), the cost of a new drug in Germany that will be covered by the SHI funds is then determined in price negotiations between the SHI bodies and the pharmaceutical company. The extent of the added benefit is graded in ascending order by
- minor (lowest extent),
- considerable (medium extent) and
- major (highest possible extent).
If the assessment does not show an added benefit (either because it is not proven in the studies or because the data are insufficient for a meaningful comparison), the drug can remain on the market. However, the cost will only be covered at the reference price set for this group of drugs or at a cost (for one year of treatment) that is not allowed to be higher than the cost of standard treatment. In general, therefore, a new drug with no added benefit is not allowed to cost more than standard treatment.
The fact that no added benefit has been proven does not necessarily mean that the new price is necessarily lower than the price previously set by the pharmaceutical company if the comparator drug was already able to achieve a high price due to patent protection.
Current and past benefit assessments are documented on the G-BA website.


{kind=link}
{kind=link}