Europäische Nutzenbewertung
In Deutschland ist die frühe Nutzenbewertung von Arzneimitteln seit 2011 etabliert und wird für (fast) alle neuen Arzneimittel und Indikationen durchgeführt. Sie dient als Basis für die Preisverhandlungen, schafft Transparenz zur Studienlage und ist bei allen Akteuren seit Jahren etabliert und akzeptiert.
Zwischen den europäischen Ländern allerdings bestehen Unterschiede bei der Bewertung von Arzneimitteln nach der Zulassung für Erstattungsentscheidungen, etwa im Bewertungsumfang, dem Vergleichsmaßstab oder dem Entscheidungshorizont. Dies ist bedingt durch unterschiedliche Gesundheitssysteme, Finanzierungen und Rechtsgrundlagen der einzelnen Länder.
Um die Qualität und Effizienz der Nutzenbewertung innerhalb der Mitgliedstaaten der EU zu fördern, wurde mit der Verordnung (EU) 2021/2282 eine gesetzliche Grundlage für die gemeinsame Nutzenbewertung geschaffen. Diese Verordnung stellt die Grundlage für die europäische Zusammenarbeit im Bereich Health Technology Assessment (HTA) dar. Die Themen dieser Zusammenarbeit sind:
- Gemeinsame klinische Bewertung (JCA – Joint Clinical Assessment) von Arzneimitteln und Medizinprodukten (Klasse IIb, III) / In-vitro-Diagnostika (Klasse D)
- Gemeinsame wissenschaftliche Beratungen (JSC – Joint Scientific Consultations)
- Ermittlung neu entstehender Gesundheitstechnologien (EHT – Emerging Health Technologies)
- Freiwillige Zusammenarbeit (z. B. zur Bewertung nicht-klinischer Aspekte wie die Bewertung der Kosten)
Die jetzt formalisierte europäische HTA-Zusammenarbeit entwickelte sich aus vormals freiwilligen Strukturen. So wurden zwischen 2006 und 2021 erste gemeinsame wissenschaftliche Standards erarbeitet, wenn auch noch ohne Rechtsverbindlichkeit. Das IQWiG hat sich bereits an dieser Phase der Zusammenarbeit intensiv beteiligt.
Seit Beginn des Jahres 2025 werden die europaweiten Health-Technology-Assessments (EU-HTA) gestaffelt eingeführt: Im Januar 2025 fiel der Startschuss für onkologische Wirkstoffe und Arzneimittel für neuartige Therapien, die ATMPs. 2028 kommen Orphan Drugs hinzu, und ab 2030 werden alle neu zugelassenen Arzneimittel auf europäischer Ebene gemeinsam bewertet. Ab 2026 sollen auch ausgewählte Medizinprodukte mit hohem Risiko europaweit bewertet werden.
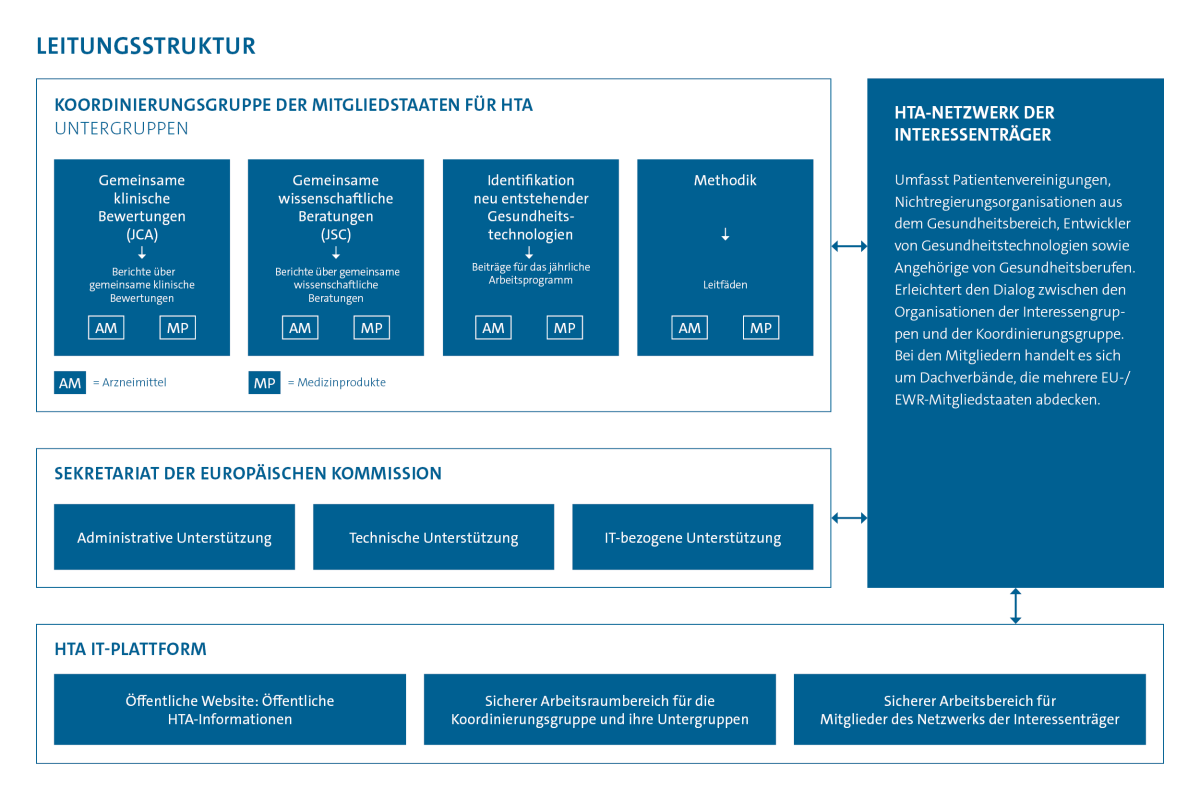
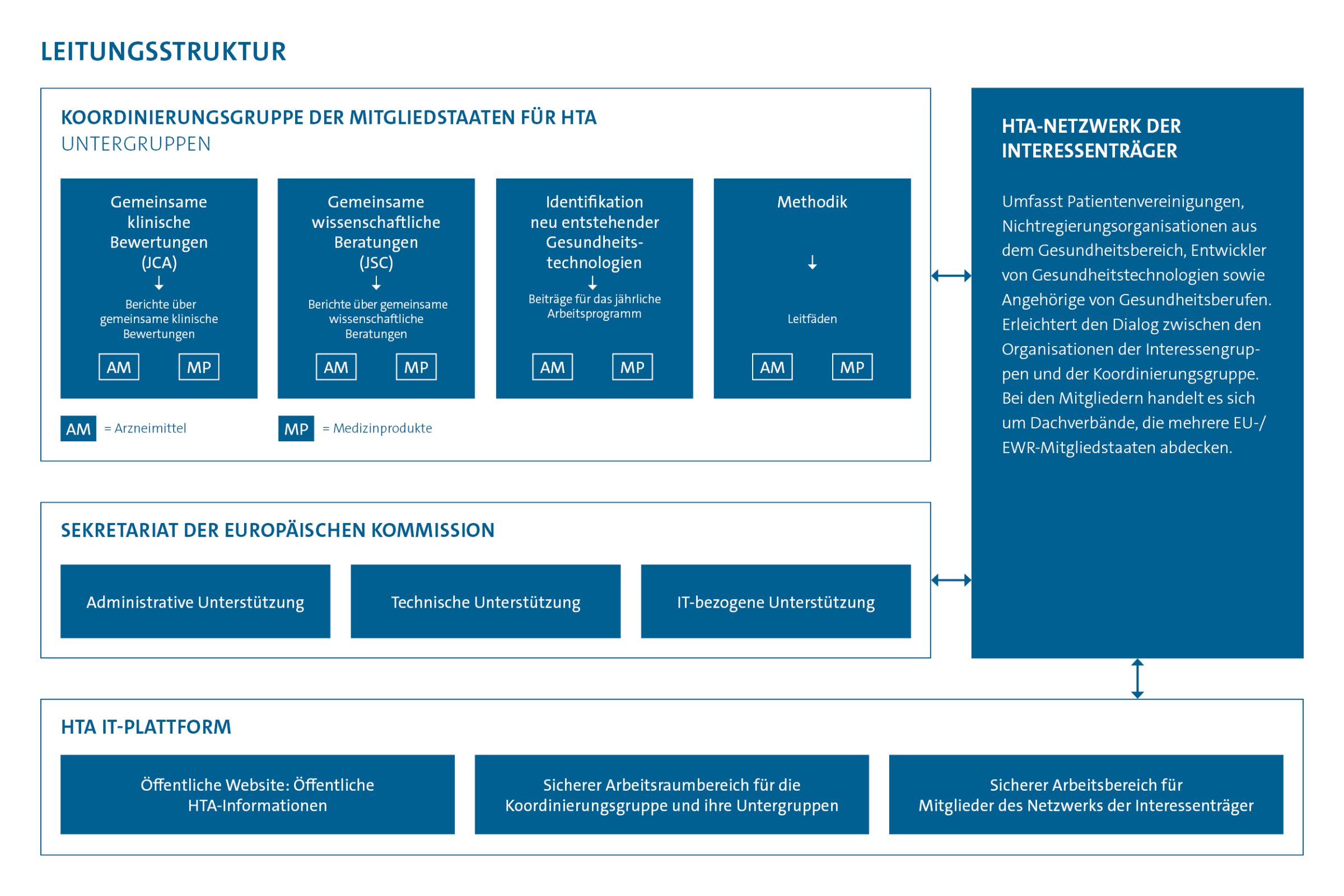
Wie ist Deutschland vertreten?
Deutschland ist in der HTA-Koordinierungsgruppe durch das Bundesgesundheitsministerium (BMG), den Gemeinsame Bundesausschuss (G-BA) und das IQWiG vertreten. In den Untergruppen wirken der G-BA und das IQWiG. Das besondere deutsche Engagement für das EU-Verfahren zeigt sich durch die Leitung der JSC-Untergruppe durch eine Vertreterin des G-BA und die Leitung der Methodik-Untergruppe durch eine Vertreterin des IQWiG.
Weitere Information zum Hintergrund finden sich auf den Internetseiten der Europäischen Kommission.
Joint Clinical Assessments (JCA)s
Gemeinsame klinische Bewertungen (Joint Clinical Assessments, JCAs) bilden den Kern der europäischen HTA-Aktivität. Die Bewertung eines neuen Arzneimittels im Vergleich mit dem bisherigen Behandlungsstandard erfolgt dabei im Rahmen vorab definierter Fragestellungen. Diese Fragestellungen werden als „PICOs“ oder „assessment scope“ bezeichnet.
„PICO“ ist ein Akronym für Population, Intervention, Comparator (= Vergleichstherapie) und Outcome (= gesundheitsbezogene Endpunkte). Ein PICO soll die Fragestellung in den jeweiligen Mitgliedsländern widerspiegeln; es ist deshalb möglich, dass Mitgliedsstaaten verschiedene Fragestellungen definieren. Für die Bestimmung ist unter anderem der rechtliche Kontext, die jeweilige Versorgungspraxis und die Verfügbarkeit von Vergleichstherapien relevant. So kann es sein, dass ein Arzneimittel, das in Deutschland als Komparator infrage kommt, in einem anderen Land nicht erstattet und daher nicht für das PICO bestimmt werden kann. In Konsequenz kann es zur Bestimmung mehrerer PICOs für die europäische Bewertung kommen.
An einem JCA arbeiten immer zwei HTA-Agenturen aus zwei europäischen Ländern – als Assessor und Co-Assessor. Diese gemeinsame Bearbeitung umfasst sowohl die Festlegung des Assessments Scopes als auch die Evidenzbewertung. Das IQWiG als erfahrene HTA-Organisation arbeitet bereits als Assessor bzw. Co-Assessor an JCAs.
| Nummer | Projekttitel | Wirkstoff | Kurzfassung Anwendungsgebiet | Gebiet | Partner | Start | vorauss. Abschluss |
|---|---|---|---|---|---|---|---|
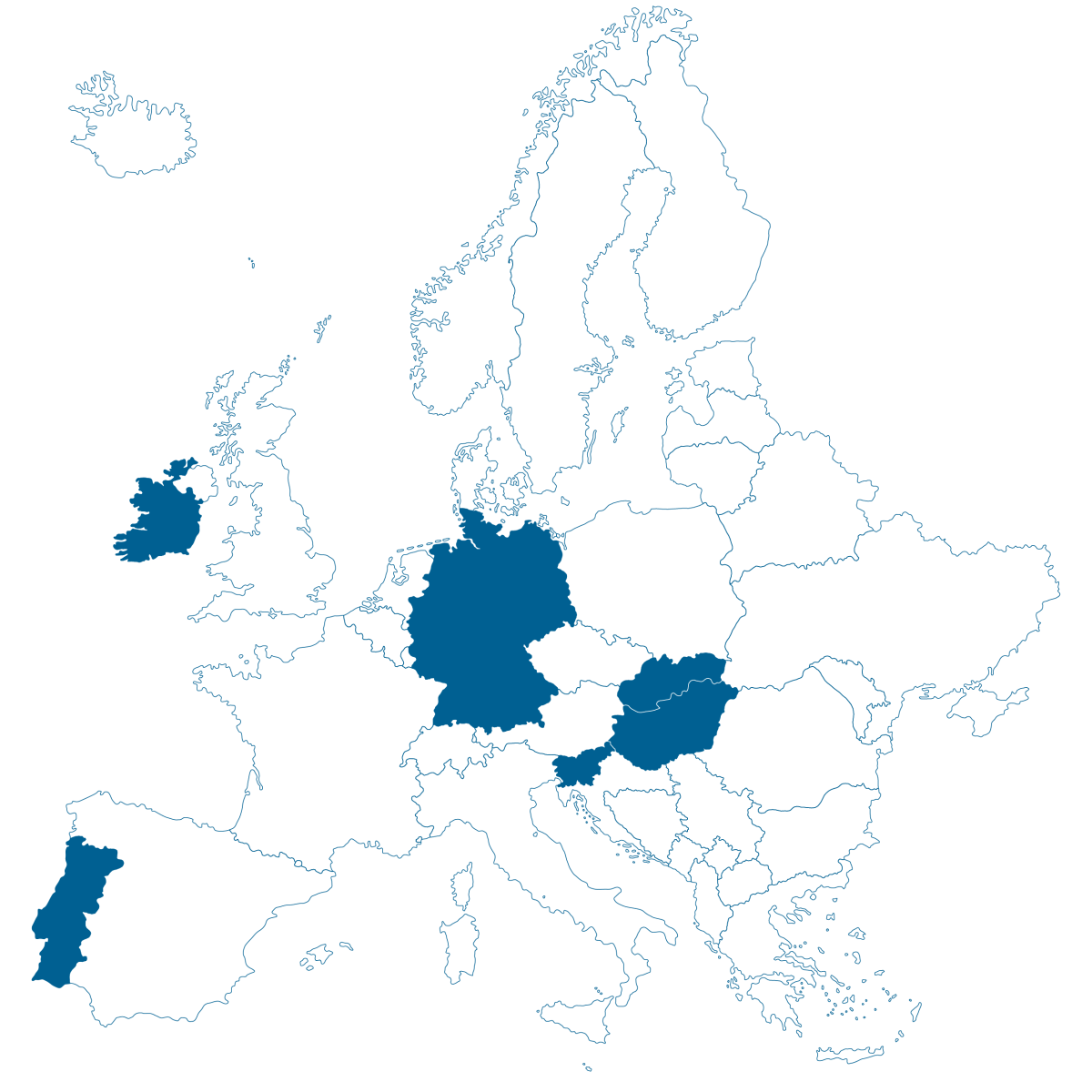
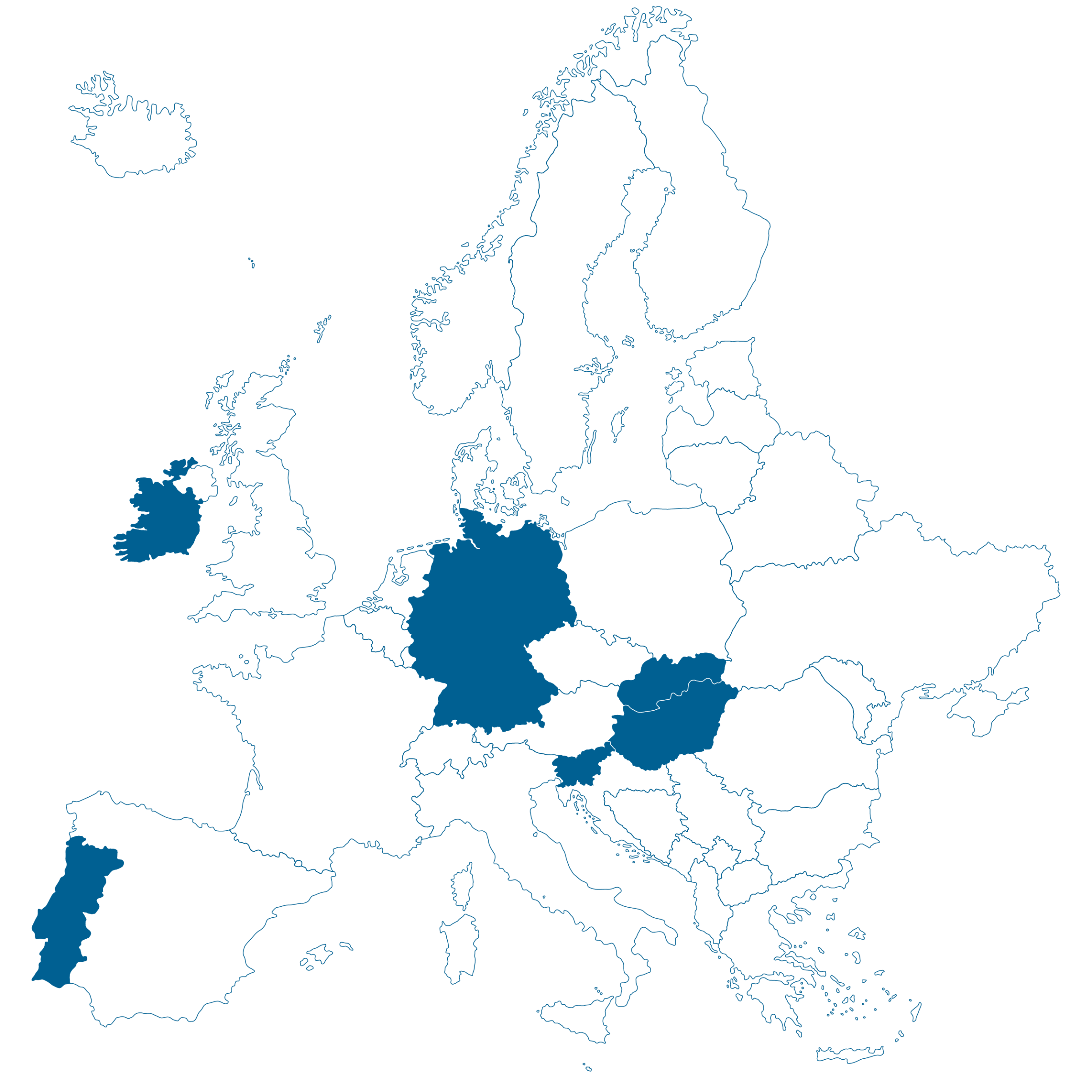
| MP-202406 | Tovorafenib (Gliom) | Tovorafenib | Behandlung des pädiatrischen niedriggradig malignen Glioms | Onkologie | NCPE (National Centre for Pharmacoeconomics), Ireland |
März 2025 | Q2 2026 |
| MP-202416 | Lurbinectedin (SCLC) | Lurbinectedin | Erhaltungstherapie beim kleinzelligen Lungenkarzinom im fortgeschrittenen Stadium (ES-SCLC) | Onkologie | Infarmed (National Authority of Medicines and Health Products), Portugal | Juni 2025 | Q3 2026 |
| MP-202417 | Tarlatamab (SCLC) | Tarlatamab | Behandlung des kleinzelligen Lungenkarzinoms im fortgeschrittenen Stadium (ES-SCLC) | Onkologie | NCPHP (National Centre for Public Health and Pharmacy), Hungary | Juni 2025 | Q3 2026 |
| MP-202418 | Senaparib (Ovarialkarzinom) | Senaparib | Erhaltungstherapie beim Ovarialkarzinom, Eileiterkarzinom oder primäres Peritonealkarzinom | Onkologie | Slovenian Quality Health Care Agency | Juli 2025 | Q3 2026 |
| MP-202508 | Sintilimab (NSCLC) | Sintilimab | Behandlung des nicht-kleinzelligen Lungenkarzinom (NSCLC) | Onkologie | National Institute for value and technology in health (NIHO), Slovakia | November 2025 | Q4 2026 |
Diese Übersicht zeigt, mit welchen Ländern das IQWIG als Assessor oder Co-Assessor zusammenarbeitet:
{kind=link}
{kind=link}
Bedeutung für das AMNOG Verfahren in Deutschland
Die JCAs sollen nationale Entscheidungen vorbereiten und unterstützen, sie aber nicht ersetzen. Die JCAs beschreiben ausschließlich die verfügbare Evidenz, also die relativen Effekte und deren Aussagesicherheit. Die Einschätzung des Zusatznutzens neuer Arzneimittel erfolgt weiterhin auf nationaler Ebene. Liegt ein europäisches Dossier vor, kann der pharmazeutische Unternehmer im deutschen Verfahren darauf verweisen, ohne die Unterlagen erneut einreichen zu müssen. Die Mitgliedsstaaten dürfen jedoch zusätzliche klinischen Analysen für den nationalen HTA-Prozess anfordern und durchführen.
Für Deutschland bedeutet dies, dass nach dem JCA noch eine Nutzenbewertung im AMNOG-Verfahren erfolgt, einschließlich einer Beschlussfassung durch den G-BA. Denn mit den Anpassungen der rechtlichen Grundlagen in der Arzneimittel-Nutzenbewertungsverordnung wurde klargestellt, dass die Kernelemente des nationalen Verfahrens – Transparenz, Möglichkeit zur Stellungnahme, Fristentreue, zweckmäßige Vergleichstherapie, Bewertungsmaßstäbe – unverändert bleiben.
Durch die schrittweise Einführung der JCA auf europäischer Ebene entsteht in Deutschland ein Nebeneinander von JCAs mit anschließender nationaler Bewertung und rein nationalen Bewertungen. Zudem werden alle Wirkstoffe, die bereits national bewertet worden sind und für die ein neues Anwendungsbiet zugelassen wird, weiterhin ausschließlich national bewertet. Dieses Nebeneinander wird voraussichtlich für 15 Jahre andauern. Damit es fair bleibt, ist es entscheidend, dass die Bewertungsmaßstäbe in beiden Fällen übereinstimmen.